Craniosynostosis is a condition that affects the normal development of a child’s skull. It occurs when one or more sutures in an infant’s skull close prematurely, potentially leading to abnormal head shapes and, in some cases, increased intracranial pressure or developmental delays. Understanding this condition is crucial for early detection and intervention, which can significantly improve health outcomes. This article provides a comprehensive overview of craniosynostosis, including its causes, symptoms, diagnosis, treatment options, and support strategies for affected children and their families. By exploring these aspects, readers can gain valuable insights into managing this complex condition effectively.
Craniosynostosis is a complex cranial deformity that requires careful diagnosis and management. Early intervention can greatly reduce the risk of complications and support healthy development. With advancements in surgical techniques and supportive care, children with craniosynostosis can achieve better functional and aesthetic outcomes. Raising awareness and understanding of this condition is essential for parents, caregivers, and healthcare providers to ensure timely treatment and ongoing support. Continued research and multidisciplinary approaches will further enhance the quality of life for children affected by craniosynostosis and their families.
Understanding Craniosynostosis: An Overview of This Skull Condition
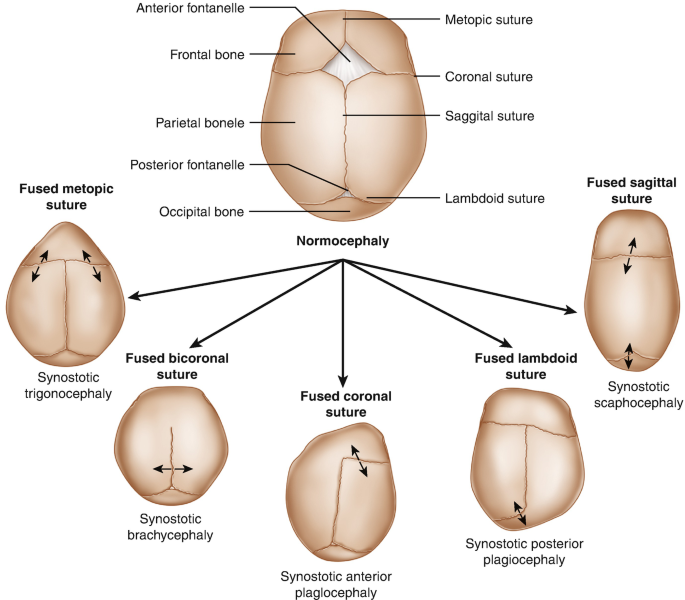
Craniosynostosis is a congenital condition characterized by the premature fusion of one or more sutures in an infant’s skull. Normally, these sutures remain open during early childhood, allowing the skull to expand as the brain grows. When they close too early, it can lead to abnormal head shapes and, in some cases, increased intracranial pressure. The condition can occur in isolation or as part of syndromic disorders involving other anomalies. Understanding the underlying mechanisms helps in recognizing the importance of early diagnosis and tailored treatment strategies. The severity and appearance of craniosynostosis can vary widely depending on which sutures are affected and the timing of suture closure.
The skull of an infant is composed of several bones connected by sutures, which are fibrous joints that allow flexibility and growth. These sutures typically remain open until late childhood or early adolescence. In craniosynostosis, the early fusion of these sutures disrupts normal skull growth, leading to characteristic skull shapes and facial features. The condition can affect any suture, with the most commonly involved being the coronal, sagittal, metopic, and lambdoid sutures. The timing of suture closure is crucial; when it occurs prematurely, it can cause asymmetrical skull deformities or increased intracranial pressure, affecting brain development.
Craniosynostosis can be classified into two main types: syndromic and nonsyndromic. Nonsyndromic craniosynostosis occurs in isolation without other abnormalities, whereas syndromic forms are associated with genetic syndromes such as Crouzon, Apert, or Pfeiffer syndrome. These syndromic cases often involve multiple sutures and additional craniofacial abnormalities. The distinction between these types influences the approach to management and prognosis. Overall, craniosynostosis is a complex condition that requires a multidisciplinary approach for accurate diagnosis and effective treatment planning.
The epidemiology of craniosynostosis indicates that it affects approximately 1 in 2,000 to 2,500 live births. It appears across all ethnic groups and both sexes, although some studies suggest a slightly higher prevalence in males. The exact cause remains unknown in many cases, but genetic factors play a significant role, especially in syndromic forms. Environmental influences and intrauterine factors may also contribute, though evidence is limited. Recognizing the early signs and understanding the diversity of presentations are essential for timely intervention, which can prevent complications and promote normal development.
Research continues to explore the genetic and environmental factors involved in craniosynostosis. Advances in molecular genetics have identified specific gene mutations associated with syndromic cases, providing insights into the pathophysiology of the condition. This knowledge assists clinicians in diagnosis and genetic counseling for affected families. Early detection and intervention are vital because untreated craniosynostosis can lead to abnormal brain growth, developmental delays, and aesthetic concerns. As such, awareness and education are key components in managing this condition effectively.
Causes and Risk Factors Associated with Craniosynostosis
The exact cause of craniosynostosis remains largely idiopathic, but several factors have been identified that increase the risk of its development. Genetic mutations are a primary cause in syndromic cases, where specific gene alterations disrupt normal suture development and fusion timing. For example, mutations in genes such as FGFR2, FGFR1, and TWIST1 are commonly associated with syndromic craniosynostosis. These genetic factors often follow inheritance patterns, making family history an important consideration in diagnosis. Understanding the genetic basis helps in early detection, counseling, and potential future therapies targeting the underlying mechanisms.
Environmental influences during pregnancy may also contribute to the risk of craniosynostosis, although evidence remains inconclusive. Factors such as maternal smoking, certain medications, or exposure to environmental toxins have been studied, but definitive links have not been established. Some researchers suggest that intrauterine constraints, such as limited space in the womb or abnormal fetal positioning, could exert mechanical forces that influence skull development. However, these are generally considered minor contributors compared to genetic factors. Overall, the multifactorial nature of craniosynostosis indicates that both genetic predispositions and environmental influences may play roles in its etiology.
Certain risk factors increase the likelihood of craniosynostosis, including a family history of cranial deformities or genetic syndromes. Advanced maternal age and multiple pregnancies, such as twins or triplets, are also associated with a higher incidence. Additionally, prenatal exposures to specific medications, such as retinoic acid, have been suggested as potential risk factors. Premature birth or low birth weight might also be linked to increased vulnerability due to disrupted fetal development. Recognizing these risk factors enables healthcare providers to monitor at-risk infants more closely for early signs of craniosynostosis.
In some cases, craniosynostosis occurs as part of broader syndromic conditions involving multiple congenital anomalies. These syndromes often involve complex genetic mutations affecting multiple developmental pathways. For example, Apert syndrome involves mutations in FGFR2 and presents with craniosynostosis, syndactyly, and other abnormalities. Such syndromic cases tend to have more severe cranial deformities and require comprehensive management. Understanding the causes and risk factors helps in differentiating isolated from syndromic craniosynostosis, guiding appropriate genetic testing and counseling for families.
Despite extensive research, many cases of craniosynostosis remain idiopathic, emphasizing the need for ongoing studies to uncover additional genetic and environmental contributors. Advances in prenatal imaging and genetic testing are improving early detection, allowing for timely intervention. Prevention strategies are limited due to the unclear etiology, but awareness of risk factors can facilitate early diagnosis and treatment. Ultimately, a combination of genetic counseling, careful prenatal care, and vigilant postnatal monitoring is essential for managing the complex causes of craniosynostosis.
Common Symptoms and Signs to Recognize in Infants
Infants with craniosynostosis often present with distinctive skull deformities that become apparent within the first few months of life. The most noticeable signs include an abnormal head shape, which varies depending on the sutures involved. For example, sagittal suture fusion results in a long, narrow skull (scaphocephaly), while coronal suture fusion can cause a short, broad skull (brachycephaly). The metopic suture fusion may produce a triangular forehead (trigonocephaly). These deformities are often symmetrical but can sometimes be asymmetrical, leading to facial asymmetry. Early recognition of these signs is vital for prompt diagnosis and management.
In addition to skull shape abnormalities, infants may exhibit a palpable ridge along the fused suture, which can be felt during physical examination. Some children may also have an increased head circumference, especially if intracranial pressure is elevated. Other signs include a prominent forehead, asymmetrical facial features, and a bossed or ridged skull. In severe cases, signs of increased intracranial pressure such as vomiting, irritability, or developmental delays may be observed. However, these symptoms are less common in mild cases and often develop later if the condition progresses untreated.
Behavioral and developmental signs can also be indicative of craniosynostosis, especially if brain growth is affected. Infants might show delays in reaching developmental milestones like sitting, crawling, or walking. In some cases, there may be visual disturbances due to increased pressure on the orbits or optic nerves. Headaches are rare in infants but may become more apparent as the child grows older if intracranial pressure persists. Careful monitoring of growth patterns and developmental progress is essential for early detection of potential complications related to craniosynostosis.
Parents and caregivers should be aware of the subtle signs that could indicate craniosynostosis, particularly if there is a family history of cranial deformities. Routine pediatric check-ups include head circumference measurements and physical examinations that can reveal early deformities. Noticing an unusual head shape, ridge along sutures, or asymmetry should prompt further assessment by a specialist. Early detection allows for timely imaging and intervention, minimizing the risk of complications and supporting normal development.
In some cases, craniosynostosis may be associated with other congenital anomalies, such as facial asymmetry, ear anomalies, or dental irregularities. These associated features can serve as additional clues for diagnosis, especially in syndromic cases. Healthcare providers often perform comprehensive evaluations to identify any related abnormalities. Imaging studies, such as cranial X-rays or CT scans, are used to confirm the diagnosis and determine which sutures are fused. Recognizing the signs early ensures that appropriate treatment plans can be devised to address both aesthetic and functional concerns.
Overall, awareness of the common symptoms and signs of craniosynostosis in infants plays a critical role in early diagnosis